EMT, Signaling Pathway & Stemness FAQs
Find scientific technical answers on frequently asked questions (FAQs):
- What is EMT and how is it significant in biosciences?
- How is EMT different from de-differentiation of the cultured cells?
- What are the key signaling pathways involved in EMT regulation?
- How do transcription factors regulate EMT?
- What are the most important EMT markers?
- Is it possible to perform a quantitative analysis of EMT?
- Can you suggest any preliminary in vitro assays for detecting EMT before I study the molecular markers?
- How can I analyze EMT in cancer xenograft tissues from control and drug-treated mice?
- I am running IHC on frozen tissue sections to determine alpha-SMA expression. Do I need to perform antigen retrieval?
- How can I increase the expression of E-cadherin and ZEB1/ZEB2 in my Western blots?
- What is an ideal housekeeping gene/protein for use as a control in EMT studies?
- Do you offer any EMT-inducing cell culture medium?
- Does Bio-Techne offer any small molecules for induction of stem cell differentiation?
- Why do cancer cells undergo EMT and acquire increased stemness?
- What are cancer-associated fibroblasts (CAFs) and how do they affect EMT?
- How does hypoxia impact EMT in cancers?
- How do exosomes facilitate EMT in cancers?
- Is there any link between autophagy and EMT or stemness?
What is EMT and how is it significant in biosciences?
Epithelial-Mesenchymal Transition (EMT) refers to the trans-differentiation of stationary epithelial cells into motile mesenchymal cells. Major changes which occur during EMT are the down-regulation of epithelial markers (such as E-Cadherin, TJP1/ZO-1 and Occludin), rearrangement of the cytoskeleton, loss of cell-cell adhesion and apical-basal polarity, and the acquisition of mesenchymal phenotype markers (such as Vimentin, N-Cadherin and Fibronectin). These mesenchymal proteins confer increased cell protrusions and motility/migratory potential of the differentiated cells. EMT was initially described by Elizabeth Hay in the 1980s as ‘epithelial to mesenchymal transformation’. However, because of its transient or transitional nature, this process was renamed later on as “epithelial-mesenchymal transition”. On the other hand, Mesenchymal-Epithelial Transition (MET) is the reverse process (of EMT) which results in the differentiation of motile mesenchymal cells to stationary epithelial cell type. A coordinated interplay between EMT and MET signals is fundamental to a wide range of biological processes including primary/secondary epithelia generation during embryogenesis/organogenesis, wound healing, and also to the pathophysiology of fibrosis and malignancies. In cancer, EMT drives invasion, therapeutic resistance, stemness and cancer spread to distant sites in the body (Stone et al. 2016; Nieto et al. 2016).
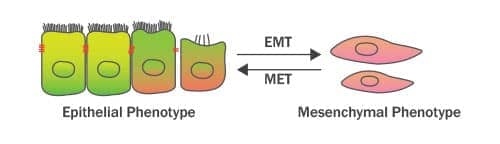
How is EMT different from de-differentiation of the cultured cells?
De-differentiation is a process whereby a specialized lineage of cells regresses to a simpler state of stem cells. Like EMT, changes in dedifferentiation may occur at the gene, protein, cell morphology/phenotype, and functional levels, but it is not limited to a single tissue type. EMT is a phenomenon exhibited by epithelial cells while de-differentiation can be induced in several different types of cells (e.g. epithelial, neural, embryonic or fibroblastic cell types). In experimental biology and regenerative medicine, de-differentiation has been extensively employed as an approach to generate pluripotent stem cells. Cells derived from different lineages, but from the same animal or subject, are often used to generate pluripotent stem cells. In addition to limiting risks of immune rejection of genetically incompatible allogenic stem cells post-transplant, this method of generating stem cells limits ethical questions raised by the use of embryonic stem cells for therapeutic and research purposes. EMT on the other hand has been of great interest in cancer biology for its usefulness in diagnosis of invasive or metastatic progression of tumors.
What are the key signaling pathways involved in EMT regulation?
EMT involves critical changes in epithelial cells that favor the development of an invasive phenotype with an increase in motility: a hallmark of mesenchymal cell populations. At the structural level, these changes include the loss of cell-cell junctions and apical-basal polarity, cytoskeletal reorganization, and alterations in expression level of proteins critical for defining cellular shape. At the molecular level, the progression of EMT is controlled by multiple signaling pathways and their coordinated interplay is critical for the induction of EMT responses. One of the most studied EMT-inducing proteins is transforming growth factor-beta (TGF-beta), which acts at various regulatory levels, and via SMAD/non-SMAD signaling. TGF-beta interacts with TGF-beta type I/II receptors to activate SMAD2/3, resulting in SMA2/3/4 complex generation. The latter translocates to the nucleus of the cell where it interacts with transcription factors responsible for controlling genes favoring EMT phenotype. Additionally, TGF beta impacts the induction of miRNAs which are known to down-regulate the expression of epithelial markers. Besides regulating SMAD and miRNAs, TGF-beta induces PI3K-AKT, ERK-MAPK, p38-MAPK and JNK signaling also for controlling EMT progression. TGF-beta type 1 receptor mediated phosphorylation of SHCA generates a docking site for GRB2 and SOS which activates RAS-RAF-MEK-ERK MAPK signaling. TGF beta receptor complex-TRAF6 interaction activates TAK1 which further induces p38 MAPK and JNK, and hence the EMT progression. Upon stimulation through several different growth factors (EGF, FGF, HGF and VEGF), receptor tyrosine kinases (RTKs) activate RAS-RAF-MEK-ERK MAPK signaling cascade. The activated ERK1/2-MAPK induces key transcription factors, as well as EMT promoting regulators of cell motility and invasion (RHO GTPases and S6K). Wnt signaling is known to drive EMT through GSK-3beta inhibition and beta-catenin stabilization. Upon its nuclear translocation, beta-catenin interacts with transcription factors TCF-LEF and activates the EMT program. RTKs or integrin induced AKT activation leads to increase in the expression of SNAI1 via inhibition of GSK-3 beta. GLI1 induces SNAI1 (Snail) expression in Hedgehog signaling, while SNAI2 (Slug) expression is up-regulated by the intracellular domain of Notch, which can lead to a decreased E-cadherin expression (a hallmark of EMT). IL6 and other inflammatory cytokines secreted by cancer cells promote EMT via STAT3-induced SNAI1/Snail expression. In tumor microenvironment, the hypoxia induced accumulation of HIF-1 alpha activates the expression of TWIST which ultimately induces EMT.
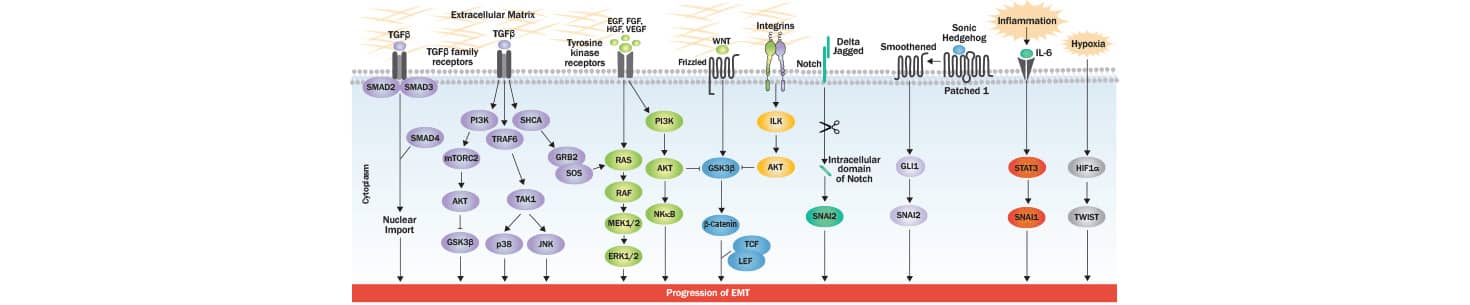
How do transcription factors regulate EMT?
At the transcriptional control level, EMT is driven by the factors: SNAI1, ZEB and basic helix–loop–helix (bHLH) proteins. These factors repress epithelial marker genes and activate genes associated with the mesenchymal phenotype. The activities of these transcription factors are regulated through their post-translational modifications, subcellular localization, and stability. Specifically, GSK-3 beta and/or PKD1 (PKC mu) phosphorylates SNAI1 which leads to its nuclear export followed by degradation via the ubiquitin (Ub)-conjugation system. SNAI1’s nuclear retention and signaling activity are promoted through its phosphorylation by PAK1 or LATS2, or its de-phosphorylation by small C-terminal domain phosphatase1 (SCP1). SNAI2/Slug, a transcriptional repressor of E-Cadherin/CDH1, on the other hand gets degraded upon p53-mediated recruitment to the MDM2 complex. Transcriptional regulator TWIST’s nuclear translocation/activity is controlled through its phosphorylation by MAPK p38, JNK and ERK. ZEB2, another transcriptional inhibitor of CDH, gets sumoylated by PRC2 and this modification leads to its translocation into the cytoplasm which reduces its activity (Lamouille et al. 2014). The genes/proteins which get repressed (down-arrow) or up-regulated (up-arrow) through transcriptional regulators of EMT are shown below.
| SNAIL Factors | bHLH Factors | ZEB Factors | |||
|---|---|---|---|---|---|
| Repressed | Upregulated | Repressed | Upregulated | Repressed | Upregulated |
| CLDN (s) | FN1 | CLDN (s) | FN1 | TJP1 | VTN |
| OCLN | VTN | OCLN | VTN | CDH1 | CDH2 |
| CDH1 | CDH2 | CDH1 | CDH2 | PKP(s) | MMP(s) |
| DSP | COL(s) | DSP | SPARC | CRB3 | |
| PKP(s) | MMP(s) | PKGB | ITGA5 | ||
| KRT(s) | ZEB | ||||
What are the most important EMT markers?
EMT markers may be broadly classified into two sub-categories: epithelial markers and mesenchymal phenotype (stemness) markers. Some of the most important markers for these phenotypes are:
| Epithelial Markers | Mesenchymal Markers | ||
|---|---|---|---|
| E-Cadherin | Syndecan | N-Cadherin | Snail |
| Cytokeratin | MUC1 | Vimentin | Slug |
| TJP/ZO1 | Desmoplakin | Fibronectin | ETS1 |
| Laminin-1 | COL4A1 | OB-Cadherin | alpha-SMA |
| Entactin | Claudins | Vitronectin | Twist |
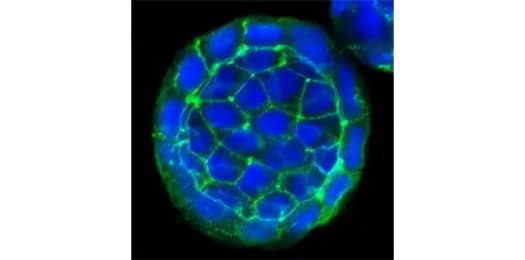
E-Cadherin Antibody (7H12) [NBP2-19051]
ICC/IF staining of E-Cadherin protein in cultured human colon cancer spheres using NBP2-19051 with DyLight 488 conjugated secondary antibody (green). The nuclei of the cells were counterstained using DAPI (blue). Note the specific staining of E-Cadherin localized to the cell membranes of the cancer cells.
![Vimentin Antibody (280618) [MAB2105]](https://resources.bio-techne.com/bio-techne-assets/images/applications/vimentin-mab2105-immunocytochemistry-12366-v3.jpg)
Vimentin Antibody (280618) [MAB2105]
ICC/IF staining of Vimentin in rat cortical stem cells using MAB2105 with NorthernLights™ 557-conjugated secondary antibody (red; Catalog # NL013) and counterstaining using DAPI (blue). Note the specific staining of Vimentin localized to the cytoskeleton.
Is it possible to perform a quantitative analysis of EMT?
Yes, one of the most commonly employed approaches is to quantify changes in the expression of molecular markers responsible for epithelial and mesenchymal phenotypes. WB, Flow cytometry, ICC/IF, and IHC are some of the assays which are useful for immuno-analysis of EMT markers. It is advisable to include 2-4 targets for both the epithelial markers (E-cadherin, pan-cytokeratin, TJP1/ZO-1, laminin-1, COL4A1, MUC1, etc.) and mesenchymal markers (N-cadherin, Vimentin, Tenascin C, alpha-SMA, F-actin, SPARC, etc). For mechanistic studies, regulators of EMT, such as SIP1, SNAI1/Snail, SNAI2/Slug, Twist1, FOXC2, and ZEB1/2 may also be analyzed.
Can you suggest any preliminary in vitro assays for detecting EMT before I study the molecular markers?
There are some assays that you can use to study the migratory and invasive potential of cells which is linked to the mesenchymal character. The most commonly used assays to assess EMT are: wound healing assays, migration chamber assays, matrigel invasion assays, gelatin zymography, and aggregation and adhesion assays. Spindle Index analysis is a relatively new assay which involves the analysis of the ratio of maximum length to maximum width of cells.
How can I analyze EMT in cancer xenograft tissues from control and drug treated mice?
In tumor xenograft tissues from various treatment groups, you can analyze EMT by using various immunoassays and gene expression/transcriptional analysis. One of the most reliable way of analyzing EMT would be to use an in situ approach, such as IHC of paraffin or frozen sections for semi-quantitative analysis of epithelial (E-cadherin, pan-cytokeratin, TJP1/ZO-1, laminin-1, COL4A1, MUC1 etc.) and mesenchymal protein markers (N-cadherin, Vimentin, Tenascin C, alpha-SMA, F-actin, SPARC etc.). Similarly, you may employ in situ hybridization (ISH) or mRNA expression studies for the transcriptional analysis of these markers. Additionally, you may isolate the cells from the tissues for ICC or flow cytometry analysis, or may prepare tissue lysates for Western blot, IP or ChIP analysis of EMT marker proteins.
![MUC-1 Antibody (SM3) [NB120-22711]](https://resources.bio-techne.com/bio-techne-assets/images/applications/muc1Left.jpg)
MUC-1 Antibody (SM3) [NB120-22711]
IHC-P analysis of a formalin fixed tissue section of human breast cancer xenograft using MUC-1 antibody clone SM3. The xenograft section depicted a very specific and intense signal in the periphery of the cancer cells. The necrotic cells also developed a strong immuno-positivity while the tumor stroma as well as the nuclei of cells did not develop any signal.

alpha-SMA Antibody (1A4) [MAB1420]
ICC/IF staining of BG01V human embryonic stem cells differentiated cardiomyocytes using alpha SMA antibody (clone 1A4) and NorthernLights™ 557-conjugated anti-mouse IgG secondary antibody (red; NL007). Nuclei were counterstained with DAPI (blue). Specific staining was localized to cytoplasm and cytoskeleton.
I am running IHC on frozen tissue sections to determine alpha-SMA expression. Do I need to perform antigen retrieval?
It is a common misconception that antigen retrieval is optional when performing IHC on frozen sections. Actually, the requirement for an antigen retrieval step in immunostaining protocols is primarily dependent upon the nature of the fixative used and the fixation time. In situations wherein the tissues are fixed in formalin/para-formaldehyde solution for more than a few hours, an antigen retrieval step is mandatory before probing the sections with primary antibody. The underlying reason is that formalin cross-links the antigenic proteins in tissues or cells hindering the antigen-antibody reaction. Without antigen retrieval, protein expression may appear low or negligible in formalin fixed tissues. For frozen sections from tissues fixed for >4 hours in formalin, we generally recommend antigen retrieval with 10 mM sodium citrate buffer (pH 6.0) for 5-10 minutes at 90 - 100 °C.
How can I increase the expression of E-cadherin and ZEB1/ZEB2 in my Western blots?
E-cadherin and ZEB1/ZEB2 are high molecular weight proteins. If weak expression is observed, then we suggest reviewing your transfer protocol from gel to membrane, as transfer of high molecular weight protein is often inefficient. Increasing the transfer time or running the transfer at 4C overnight at relatively low voltage can enhance the transfer of high molecular weight proteins on to the membrane. Additionally, you may supplement your transfer buffer with SDS (not more than 0.1% though) because SDS helps to improve transfer when the protein of interest is larger than 100kD in size. Besides optimizing the transfer step, make sure to use 6% or 8% gel for the separation of the proteins. You may also check the dilutions and incubation times for the primary and secondary antibodies. Besides technical aspects, you should investigate the nature of the samples also: E-Cadherin is expected to be expressed more in epithelial cell types while ZEB1/ZEB2 demonstrates higher expression in mesenchymal or stem cells.
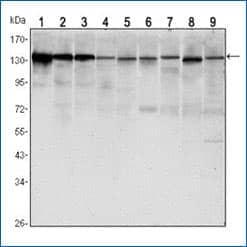
E-Cadherin Antibody (7H12) [NBP2-19051]
Western blot analysis of lysates from LNCAP (1), A431 (2), DU145 (3), PC-3 (4), MCF-7 (5), PC-12 (6), NIH/3T3 (7), C6 (8) and COS7 (9) cell lines using E-Cadherin antibody (clone 7H12) with HRP-ECL detection.
![ZEB1 Antibody [NBP2-13159]](https://resources.bio-techne.com/bio-techne-assets/images/applications/ZEB1AB.jpg)
Western blot analysis of ZEB1 in lysates from HeLa (1), COS7 (2), HepG2 (3), and Ntera2 (4) cells using rabbit polyclonal ZEB1 antibody with HRP-ECL detection. The expression of ZEB1 was found to be highest in HeLa and lowest in Ntera2 cells among the tested lines.
What is an ideal housekeeping gene/protein for use as a control in EMT studies?
Because EMT is a process which involves critical changes in the molecular expression, as well as the structural organization of various proteins in the cells, some researchers find it concerning to use structural proteins such as beta-Actin or CD44 as controls in immuno-blotting or immuno-staining analyses. GAPDH is a protein that is commonly used as a control in EMT studies, however, the expression of this protein may change with hypoxia which makes it unsuitable as a control when working on hypoxic samples. Tubulin, PARP, or other proteins, such as total-EGFR and pan-AKT, are some other options which are useful for loading control purpose. When working on Western blots, staining of the blots with Ponceau S or Amido black stain may also be used to normalize the protein.
Do you offer any EMT-inducing cell culture medium?
Bio-Techne offers StemXVivo EMT Inducing Media Supplement (100X) (Catalog #CCM017) which is useful to induce EMT in vitro in less than a week (5 days). This medium contains a mixture of EMT-inducing recombinant proteins (Wnt-5a and TGF-β1) and neutralizing antibodies (anti- E-Cadherin, anti- sFRP-1 and anti-Dkk-1) for induction of EMT. This product has a defined composition, which gives consistent results and it is compatible with a range of cell lines such as MCF-7, MCF-10A, HT-29, A549 and A431.
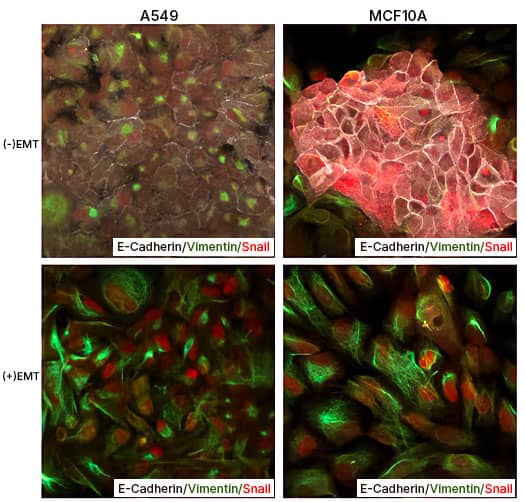
EMT Induction in A549 and MCF 10A cells with StemXVivo EMT Inducing Media Supplement (100X) Human lung carcinoma cells A549 and breast epithelial cells MCF 10A were either left untreated or were treated with the StemXVivo™ EMT Inducing Media Supplement (Catalog # CCM017) for 5 days. Thereafter, the cells were studied for EMT induction using Human EMT 3-Color Immunocytochemistry Kit (Catalog # SC026) which allows the simultaneous/single step fluorescent immunostaining of 3 EMT markers. In the data image, note the staining of E-Cadherin (white), Snail (red), and Vimentin (green).
Does Bio-Techne offer any small molecules for induction of stem cell differentiation?
We provide a large range of small molecules which are useful for differentiating stem cells into various cell types.
| Small Molecule | Biological Activity |
|---|---|
| AICAR (2840) | Promotes osteogenic differentiation of bone marrow-derived MSCs |
| DAPT (2634) | Induces neuronal differentiation |
| Dorsomorphin (3093) | Promotes cardiomyocyte differentiation in mouse ESCs |
| ISX 9 (4439) | Induces neuronal differentiation of SVZ progenitors |
| Wnt-C59 (5148) | Induces differentiation of iPSCs to cardiomyocytes |
| Purmorphamine (4551) | Induces differentiation of mesenchymal progenitors into osteoblasts |
|
View more compounds for stem cell differentiation |
|
Why do cancer cells undergo EMT and acquire increased stemness?
A growing tumor creates a hypoxic microenvironment to promote its own growth. It does this through the secretion of stress-related nutrients and growth factors, which prevent immune recognition and destruction. In response to the tumor microenvironment, cancer cells undergo EMT and acquire a mesenchymal phenotype and stem cell-like features. A mesenchymal or stemness-rich phenotype (cancer stem cells/CSCs) promotes tumor cell evasion and invasion/metastasis of secondary sites which are rich in nutrients, growth factors, and oxygen.
What are cancer-associated fibroblasts (CAFs) and how do they affect EMT?
In solid tumors, CAFs form a major component of the tumor stroma. Cancer cells activate the normal stromal fibroblasts (NSFs) into CAFs via the stimulation of paracrine growth factors. CAFs play a critical role in tumor growth and angiogenesis, and they are known to stimulate cancer cell invasiveness via secretion of various cytokines/factors, such as TGF-β1, PDGF, TNFα and IL-6. In comparison to NSFs, CAFs are more effective in promoting cancer cell survival, growth and progression. Importantly, some of the growth factors released from CAFs, especially TGF-β1 and CXCL12/SDF-1, are well-known EMT inducers in cancer cells (Zhuang et al. 2015).
How does hypoxia impact EMT in cancers?
Inadequate or low levels of oxygen, also known as hypoxia, is a hallmark of tumor generation. Hypoxia in the tumor microenvironment develops in growing tumors as a pathophysiologic outcome of disrupted microcirculation. Hypoxia induces the expression of hypoxia-inducing factors (HIFs), and through their target genes, HIFs regulate several signaling proteins and pathways, which in turn regulate tumor growth, metabolism, angiogenesis, EMT and stemness, and metastasis. HIF-1 alpha, the master regulator of hypoxia, directly or indirectly controls the expression of several transcriptional regulators of EMT (Twist1, Snail, Slug, ZEB1/2, and E12/E47) and of pro-EMT/stemness proteins, including TGF-beta, beta-catenin, Wnt and Notch (Kao et al. 2016).
How do exosomes facilitate EMT in cancers?
Exosomes are small membrane-enclosed vesicles which are generated during the maturation of endosomes as intra-luminal vesicles of multi-vesicular bodies. Exosomes are released into the extracellular environment via exocytosis and are now known to act as extracellular signalosomes, facilitating intercellular communication. In cancers, tumor-derived exosomes (TDEs) carry pro-EMT molecules such as TGF-β, caveolin-1, HIF-1α, and β-catenin. Upon reaching the recipient cells, these molecules trigger the generation of mesenchymal phenotype, stromal remodeling, and the formation of the pre-metastatic niche (Syn et al. 2016).
Is there any link between autophagy and EMT or Stemness?
Yes, there has been a growing interest and experimental evidence in support of a relationship between autophagy and EMT, as well as the regulation of stemness. TGF-β1 is known to induce both autophagy and EMT. Inhibition of autophagy has been shown to reverse EMT. Furthermore, a recent study showed that the autophagic disruption of E-cadherin/β-catenin complex promotes EMT through pTyr654-beta-catenin/p-SMAD2 mediated ILK induction (Pang et al. 2016). In another study involving Cadherin-6 (CDH-6), a type II cadherin responsible for EMT in embryonic development and known to be aberrantly expressed in cancers, Gugnoni et al ,2016 reported the direct interaction of CDH-6 with GABARAP, BNIP3 and BNIP3L. This interaction promoted EMT and cancer metastasis by inhibiting autophagy. Similarly, several recent studies have shown that the autophagic cell death mechanism is involved in the self-renewal and maintenance of stem cells (especially their undifferentiated states/stemness) and also contributes to stem cell differentiation (Wang et al. 2015). The relationship between autophagy and EMT is a rapidly growing research sub-area, and more studies employing small molecule or peptide inhibitors and inducers of autophagy (such as tat-Beclin D11) are warranted for a complete understanding of the interplay between autophagy and EMT signaling.
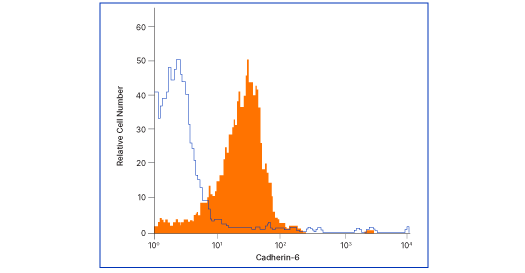
Cadherin-6/KCAD Antibody (427909) [MAB2715]
Flow analysis of MG‑63 human osteosarcoma cells with Cadherin‑6 antibody (MAB2715, filled histogram) or isotype control (# MAB002, open histogram), followed by Allophycocyanin-conjugated anti-mouse IgG F(ab')2 secondary antibody (# F0101B). Cells were stained in a buffer containing Ca2+ and Mg2+ ions.
![Tat-Beclin 1 D11 Autophagy Inducing Peptide [NBP2-49888]](https://resources.bio-techne.com/bio-techne-assets/images/applications/TatBeclin.jpg)
Tat-Beclin 1 D11 Autophagy Inducing Peptide [NBP2-49888]
HeLa GFP-LC3B cells were treated with 10uM of Tat-D11 for 1.5 hours. Thereafter, the cells were stained using NeuroTrace Red or DAPI and were analyzed employing fluorescent microscopy. Note the autophagosomes/GFP-LC3B+ puncta in the staining images of cells.
-
Stone RC, Pastar I, Ojeh N et al. (2016) Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. PMID: 27461257.
-
Nieto MA, Huang RY, Jackson RA et al. (2016) EMT Cell PMID: 27368099.
-
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition Nat Rev Mol Cell Biol. PMID: 24556840.
-
Zhuang J, Lu Q, Shen B et al. (2015) TGF beta 1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep. PMID: 26152796.
-
Pang M, Wang H, Rao P et al. (2015) Autophagy links beta-catenin and Smad signaling to promote epithelial-mesenchymal transition via upregulation of integrin linked kinase Int J Biochem Cell Biol. PMID: 27177845.
-
Gugnoni M, Sancisi V, Gandolfi G et al. (2016) Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy Oncogene PMID: 27375021.
-
Wang S, Xia P, Rehm M, Fan Z. (2015) Autophagy and cell reprogramming Cell Mol Life Sci. PMID: 25572296.
-
Syn N, Wang L, Sethi G et al. (2016) Exosome-mediated metastasis: from epithelial-mesenchymal transition to escape from immune-surveillance Trends Pharmacol Sci. PMID: 27157715.
-
Kao SH, Wu KJ, Lee WH (2016) Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes J Clin Med. PMID: 26861406.